
X-RAY RUNS: Apply for Beamtime
2017 Nov 1 - Dec 21
2018 Feb 7 - Apr 3
2018 Proposal/BTR deadline: 12/1/17
2018 Apr 11 - Jun 4
2018 Proposal/BTR deadline: 2/1/18
Within every cell of the human body lies an intricate network of membrane tubules and sheets tasked with orchestrating the processing of many of the molecules necessary to keep the cell alive. It is involved in both the synthesis of these molecules and their transport to the appropriate parts of the cell. This network, called the endoplasmic reticulum (ER), employs many different types of proteins to accomplish this task. One subset of these proteins is charged with keeping the complex structure of the ER intact so that it can continue to perform its necessary function [1]. However, when this structure breaks down, it can result in diseases.
One protein, called atlastin-1, is involved in maintaining ER structure. Atlastin-1 belongs to a larger class of proteins that can use an energy molecule called GTP to do mechanical work in the cell. For atlastin, this work involves bringing two lipid membranes together and fusing them in order to build up a web-like network of tubules in the ER. To do this, two atlastin molecules on opposite membranes must bind to GTP and use its stored energy (a process called hydrolysis) to bring the membranes close together for fusion (Figure 1). When mutated, this protein is known to cause Hereditary Spastic Paraplegia (HSP) and Hereditary Sensory Neuropathy (HSN), two diseases that result from the degeneration of neurons that control muscles [2, 3]. The ultimate cause of these diseases in terms of mutations in this protein is still unclear, but new information about the mechanism by which atlastin-1 maintains ER structure is bringing scientists closer to a better understanding of these diseases and how the ER functions in general.
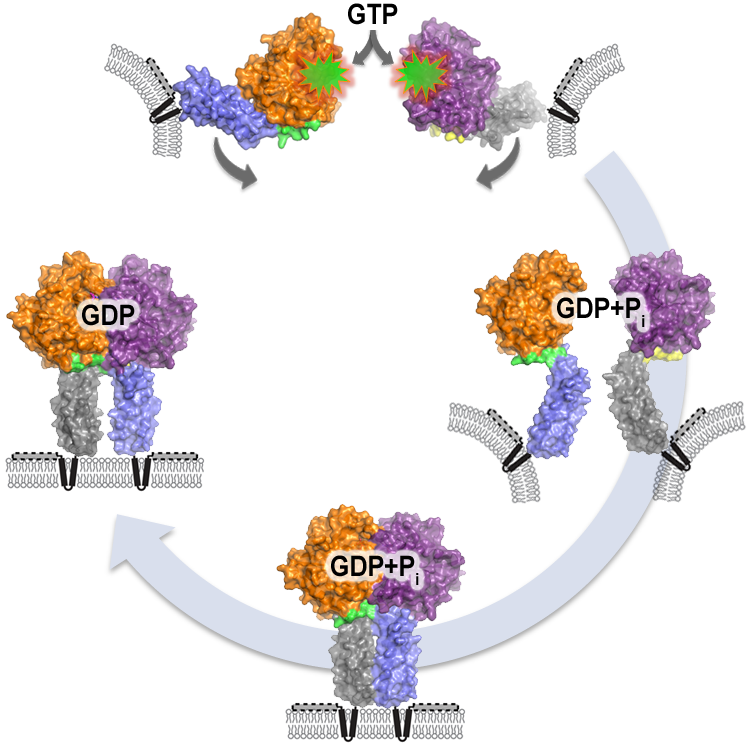
Figure 1. Model for atlastin-mediated membrane fusion. Atlastin begins in opposing membranes, with monomeric GTP-binding domains. GTP binding and hydrolysis (creating GDP + phosphate) drive a rapid conformational change between the two domains of the protein, immediately followed by dimerization of the atlastin-1 monomers. Once in this complex, membrane curvature and stress caused by atlastin’s transmembrane domains and C-terminus would allow fusion to occur spontaneously. Phosphate release follows, with relaxation and subsequent disassembly of the dimer. Other proteins that may interact with atlastin and contribute to membrane curvature are not shown.
Recently, researchers in the Sondermann group at Cornell University have uncovered new insights into the function of atlastin-1, and the results are reported in EMBO Journal [4]. They used X-ray crystallography (CHESS, beamline A1) to solve the structure of atlastin-1 bound to small molecules that mimic GTP. This showed two atlastin molecules dimerized in a tight interaction that would be able to bring opposing membranes into close proximity. They also used a technique called Förster Resonance Energy Transfer (FRET) to monitor the timing and nature of events when atlastin-1 binds to and hydrolyzes GTP. Furthermore, the work revealed how two parts of the protein work together to enable it to bind GTP and dimerize, uncovering a novel method of regulation for this type of protein. With this new information, researchers will be able to study mutations causing HSP and HSN in a more directed fashion, and possibly identify which part of the mechanism each mutation affects.
References:
[1] Shuliang Chen, Peter Novick, and Susan Ferro-Novick, (2013) ER structure and function, Curr Opin Cell Biol, doi: 10.1016/j.ceb.2013.02.006 [Epub ahead of print].
[2] Craig Blackstone, Cahir J. O’Kane, and Evan Reid (2011) Hereditary spastic paraplegias: membrane traffic and the motor pathway, Nat Rev Neurosci, 1, 31-42.
[3] Christian Guelly et al., (2011) Targeted High-Throughput Sequencing Identifies Mutations in atlastin-1 as a Cause of Hereditary Sensory Neuropathy Type I, Am J Hum Genet, 88 (1), 99-105.
[4] Laura J Byrnes, Avtar Singh, Kylan Szeto, Nicole M Benvin, John P O‘Donnell, Warren R Zipfel, and Holger Sondermann; “Structural basis for conformational switching and GTP loading of the large G protein atlastin”, EMBO J (2013), 32, 369 – 384.
Submitted by: Chae Un Kim, MacCHESS, Cornell University
4/11/2013