
X-RAY RUNS: Apply for Beamtime
2017 Nov 1 - Dec 21
2018 Feb 7 - Apr 3
2018 Proposal/BTR deadline: 12/1/17
2018 Apr 11 - Jun 4
2018 Proposal/BTR deadline: 2/1/18
Inside living cells, biological macromolecules such as proteins, RNA and DNA, are dynamic. To bridge our knowledge of static, atomic resolution structures determined using X-ray crystallography with how molecules change and interact in a more biological context, we rely on “solution structure” methods such as small angle X-ray scattering (SAXS) [1]. Although SAXS structures are low resolution, the measurement is performed on molecules that are freely diffusing: SAXS experiments may observe dynamic states and macromolecular complexes that cannot crystalize. However, the freedom that molecules have in solution comes at a price: they can aggregate, unfold, or break apart when exposed to X-rays. In fact, molecules in solution can be as much as 1000 times more sensitive to X-ray induced damage than molecules in crystals [2,3]. Because of this critical limitation, SAXS has not been able to take full advantage of high-brightness X-ray sources in order to increase throughput and reduce sample consumption.
What if you could capture molecules in their solution state, while preventing them from moving during the SAXS experiment? To find out if such an approach could reduce radiation damage in SAXS, Lois Pollack and Rob Thorne's research groups at Cornell and MacCHESS scientist Richard Gillilan borrowed a technique from crystallography called cryo-cooling. The goal of this technique is to cool the solution quickly enough that water forms a glass-like solid, rather than crystalline ice [4]. To make cryo-cooling work for SAXS, they had to leap over several technical hurdles. Unlike crystallography, for SAXS the scattering of the buffer solution must be carefully measured and subtracted to obtain the scattering from the macromolecule alone. Cryo-cooling was thought to be unsuitable for SAXS because it can cause irreproducible formation of small amounts of ice or distortions in the sample shape that would make background subtraction impossible. The Cornell team showed that by adding to their solutions sufficient amounts of a chemical similar to anti-freeze, PEG 200, small microliter-sized droplets could be rapidly cooled without forming ice (fig. 1).
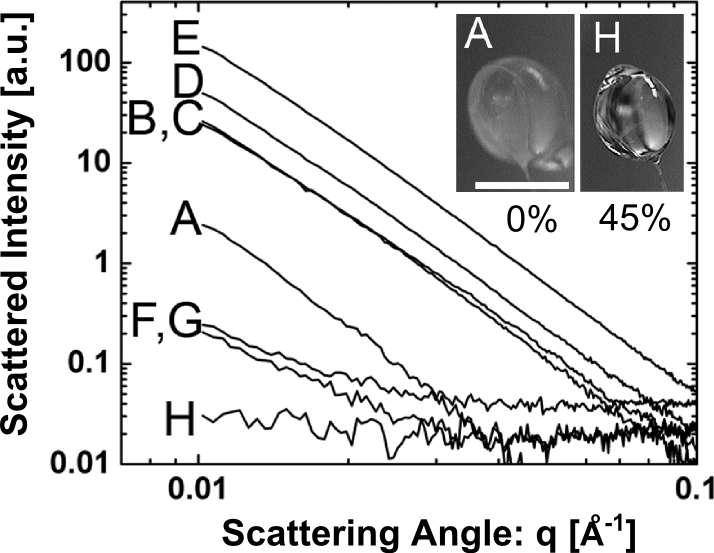
Figure 1. X-ray scattering from cryo-cooled droplets with increasing concentrations of PEG 200 (A at 0% PEG through H at 45% PEG). The characteristic power-law decay of excess scattering intensity from ice domains is evident for PEG concentrations of 40% (w/w) and below, but disappears at 45% PEG 200 (droplet H).
Although adding PEG allowed for reproducible cryo-cooling, background subtraction was still unreliable because the size of the droplets was uncontrolled. To solve this problem, the team optimized the sample mounting and developed a background subtraction method that accounts for inevitable sample thickness variations. With these improvements, they set out to validate the cryo-SAXS technique by measuring solution structures from several macromolecules at CHESS beamline G1. The low-resolution shapes obtained from the cryo-SAXS data agreed with the known atomic structures of these molecules (fig. 2).
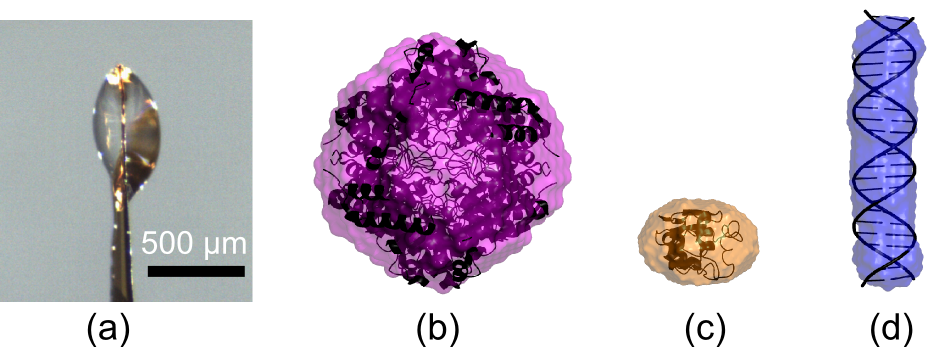
Figure 2. (a) Side view of a small droplet containing macromolecules in solution and 45% PEG 200 that was held by surface tension and rapidly cooled to 100K. Cryo-SAXS data were acquired using similarly frozen samples and matching buffers, and analyzed to yield the low resolution molecular shape. In (b-c), cryo-SAXS shape reconstructions for three macromolecules are overlayed with their known atomic structures: glucose isomerase (b), lysozyme(c), and 24 base-pair DNA (d).
Remarkably, they found that cryo-SAXS can be used with the X-ray doses that are ~ 1000 times larger, and illuminated volumes ~1000 smaller, than standard SAXS practice, with no signs of damage. These results should expand the SAXS technique to macromolecules that degrade over time, are available in limited quantity, or are especially radiation-sensitive. They also pave the way for high throughput cryo-SAXS applications involving rapid data collection and minimal sample consumption by taking advantage of high-brightness X-ray sources. Their work was published in the Biophysical Journal [5].
References:
[1] Rambo, R. P. & Tainer, J. A. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle x-ray scattering. Current opinion in structural biology 20, 128-137 (2010).
[2] Kuwamoto, S., Akiyama, S. & Fujisawa, T. Radiation damage to a protein solution, detected by synchrotron x-ray small-angle scattering: dose-related considerations and suppression by cryoprotectants. Journal of Synchrotron Radiation 11, 462-468 (2004).
[3] Southworth-Davies, R. J., Medina, M. A., Carmichael, I. & Garman, E. F. Observation of decreased radiation damage at higher dose rates in room temperature protein crystallography. Structure 15, 1531-1541 (2007).
[4] Garman, E. F. & Schneider, T. R. Macromolecular Cryocrystallography. Journal of Applied Crystallography 30, 211-237 (1997).
[5] Meisburger, S. P., Warkentin, M., Chen, H., Hopkins, J. B., Gillilan, R. E., Pollack, L., & Thorne, R. E. Breaking the radiation damage limit with Cryo-SAXS. Biophysical Journal 104, 227-236 (2013).
Submitted by: Chae Un Kim, MacCHESS, Cornell University
3/05/2013